





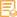


Catalog: YP0681
Size
Price
Status
Qty.
200μL
$600.00
In stock
0
100μL
$340.00
In stock
0
50μL
$190.00
In stock
0
Add to cart


Collected
Collect
Main Information
Target
XIAP
Host Species
Rabbit
Reactivity
Human, Mouse, Rat
Applications
WB, IHC, IF, ELISA
MW
57kD (Observed)
Conjugate/Modification
Phospho
Detailed Information
Recommended Dilution Ratio
WB 1:500-1:2000; IHC 1:100-1:300; ELISA 1:20000; IF 1:50-200
Formulation
Liquid in PBS containing 50% glycerol, 0.5% BSA and 0.02% sodium azide.
Specificity
Phospho-XIAP (S87) Polyclonal Antibody detects endogenous levels of XIAP protein only when phosphorylated at S87.The name of modified sites may be influenced by many factors, such as species (the modified site was not originally found in human samples) and the change of protein sequence (the previous protein sequence is incomplete, and the protein sequence may be prolonged with the development of protein sequencing technology). When naming, we will use the "numbers" in historical reference to keep the sites consistent with the reports. The antibody binds to the following modification sequence (lowercase letters are modification sites):KVsPN
Purification
The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen.
Storage
-15°C to -25°C/1 year(Do not lower than -25°C)
Concentration
1 mg/ml
MW(Observed)
57kD
Modification
Phospho
Clonality
Polyclonal
Isotype
IgG
Related Products
Primary Antibodies
XIAP Rabbit pAb
YT4913
More→
Primary Antibodies
XIAP (Phospho Ser87) Rabbit pAb
YP0681
More→
ELISA Kits

Total XIAP Cell-Based Colorimetric ELISA Kit
KA4326C
More→
Secondary Antibodies
Goat Anti Mouse IgG(H+L) (HRP)
RS0001
More→
Secondary Antibodies
Goat Anti Rabbit IgG(H+L) (HRP)
RS0002
More→
Primary Antibodies
β-actin (PTR2364) Mouse mAb
YM3028
More→
Primary Antibodies
GAPDH (PTR2304) Mouse mAb
YM3029
More→
Antigen&Target Information
Immunogen:
The antiserum was produced against synthesized peptide derived from human XIAP around the phosphorylation site of Ser87. AA range:53-102
show all
Specificity:
Phospho-XIAP (S87) Polyclonal Antibody detects endogenous levels of XIAP protein only when phosphorylated at S87.The name of modified sites may be influenced by many factors, such as species (the modified site was not originally found in human samples) and the change of protein sequence (the previous protein sequence is incomplete, and the protein sequence may be prolonged with the development of protein sequencing technology). When naming, we will use the "numbers" in historical reference to keep the sites consistent with the reports. The antibody binds to the following modification sequence (lowercase letters are modification sites):KVsPN
show all
Gene Name:
XIAP
show all
Protein Name:
E3 ubiquitin-protein ligase XIAP
show all
Other Name:
XIAP ;
API3 ;
BIRC4 ;
IAP3 ;
E3 ubiquitin-protein ligase XIAP ;
Baculoviral IAP repeat-containing protein 4 ;
IAP-like protein ;
ILP ;
hILP ;
Inhibitor of apoptosis protein 3 ;
IAP-3 ;
hIAP-3 ;
hIAP3 ;
X-linked inhibitor of apoptosis protein ;
X-linked I
API3 ;
BIRC4 ;
IAP3 ;
E3 ubiquitin-protein ligase XIAP ;
Baculoviral IAP repeat-containing protein 4 ;
IAP-like protein ;
ILP ;
hILP ;
Inhibitor of apoptosis protein 3 ;
IAP-3 ;
hIAP-3 ;
hIAP3 ;
X-linked inhibitor of apoptosis protein ;
X-linked I
show all
Database Link:
Background:
This gene encodes a protein that belongs to a family of apoptotic suppressor proteins. Members of this family share a conserved motif termed, baculovirus IAP repeat, which is necessary for their anti-apoptotic function. This protein functions through binding to tumor necrosis factor receptor-associated factors TRAF1 and TRAF2 and inhibits apoptosis induced by menadione, a potent inducer of free radicals, and interleukin 1-beta converting enzyme. This protein also inhibits at least two members of the caspase family of cell-death proteases, caspase-3 and caspase-7. Mutations in this gene are the cause of X-linked lymphoproliferative syndrome. Alternate splicing results in multiple transcript variants. Pseudogenes of this gene are found on chromosomes 2 and 11.[provided by RefSeq, Feb 2011],
show all
Function:
Disease:Defects in XIAP are the cause of lymphoproliferative syndrome X-linked type 2 (XLP2) [MIM:300635]. XLP is a rare immunodeficiency characterized by extreme susceptibility to infection with Epstein-Barr virus (EBV). Symptoms include severe or fatal mononucleosis, acquired hypogammaglobulinemia, pancytopenia and malignant lymphoma.,Domain:The first BIR domain is involved in interaction with MAP3K7IP1 and is important for dimerization. The second BIR domain is sufficient to inhibit caspase-3 and caspase-7, while the third BIR is involved in caspase-9 inhibition. The interactions with SMAC and PRSS25 are mediated by the second and third BIR domains.,Function:Apoptotic suppressor. Has E3 ubiquitin-protein ligase activity. Mediates the proteasomal degradation of target proteins, such as caspase-3, SMAC or AIFM1. Inhibitor of caspase-3, -7 and -9. Mediates activation of MAP3K7/TAK1, leading to the activation of NF-kappa-B.,online information:XIAP mutation db,PTM:Phosphorylation by PKB/AKT protects XIAP against ubiquitination and protects the protein against proteasomal degradation.,PTM:Ubiquitinated and degraded by the proteasome in apoptotic cells.,similarity:Belongs to the IAP family.,similarity:Contains 1 RING-type zinc finger.,similarity:Contains 3 BIR repeats.,subunit:Monomer, and homodimer. Interacts with SMAC and with PRSS25; these interactions inhibit apoptotic suppressor activity. Interacts with MAP3K7IP1 and AIFM1. Interaction with SMAC hinders binding of MAP3K7IP1 and AIFM1. Interacts with TCF25.,tissue specificity:Ubiquitous, except peripheral blood leukocytes.,
show all
Cellular Localization:
Cytoplasm. Nucleus. TLE3 promotes its nuclear localization.
show all
Tissue Expression:
Research Areas:
>>Platinum drug resistance ;
>>NF-kappa B signaling pathway ;
>>Ubiquitin mediated proteolysis ;
>>Apoptosis ;
>>Apoptosis - multiple species ;
>>Necroptosis ;
>>Focal adhesion ;
>>NOD-like receptor signaling pathway ;
>>Toxoplasmosis ;
>>Human T-cell leukemia virus 1 infection ;
>>Pathways in cancer ;
>>Chemical carcinogenesis - receptor activation ;
>>Small cell lung cancer
>>NF-kappa B signaling pathway ;
>>Ubiquitin mediated proteolysis ;
>>Apoptosis ;
>>Apoptosis - multiple species ;
>>Necroptosis ;
>>Focal adhesion ;
>>NOD-like receptor signaling pathway ;
>>Toxoplasmosis ;
>>Human T-cell leukemia virus 1 infection ;
>>Pathways in cancer ;
>>Chemical carcinogenesis - receptor activation ;
>>Small cell lung cancer
show all
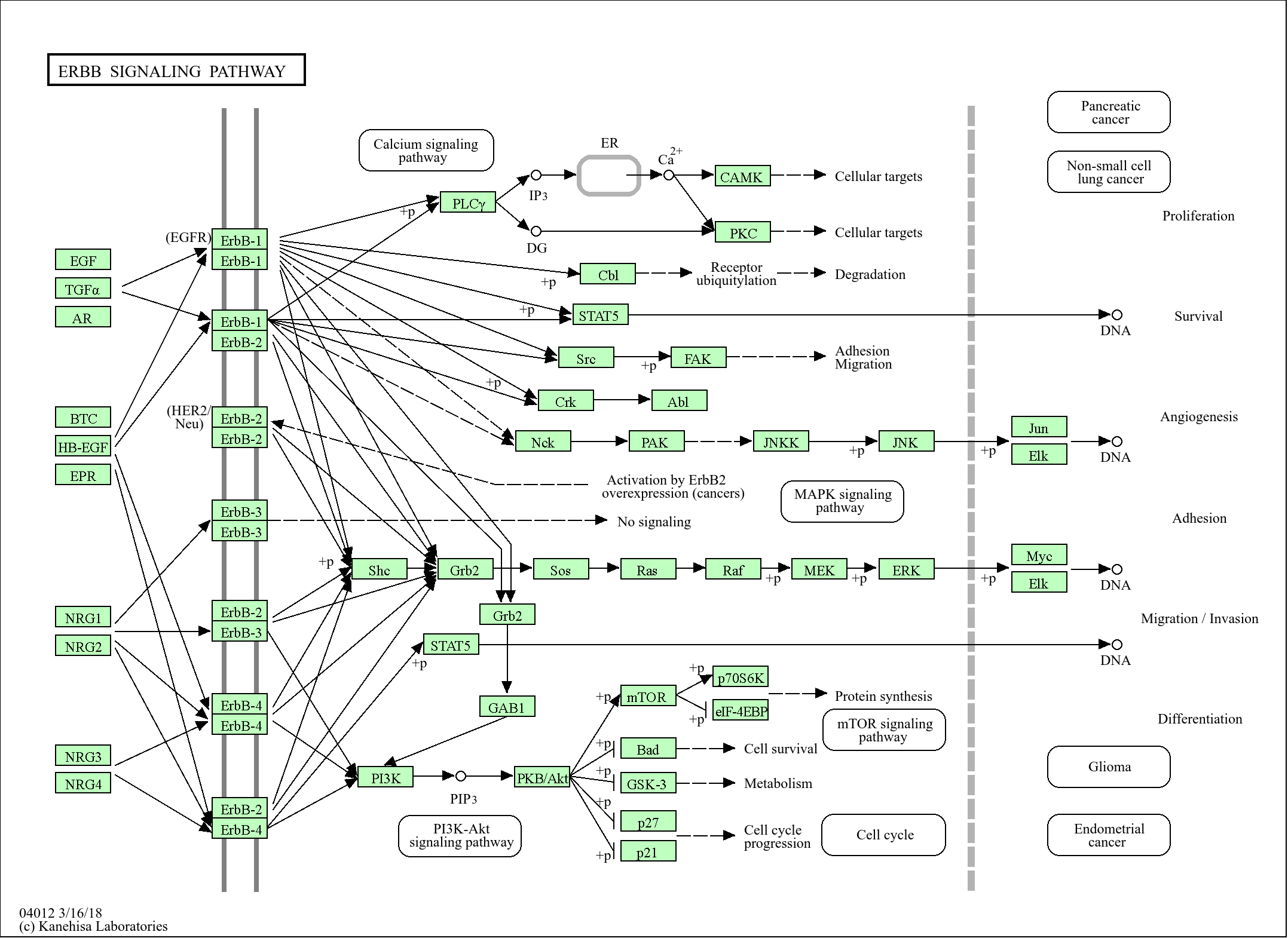
Signaling Pathway
Cellular Processes >> Cell growth and death >> Apoptosis
Cellular Processes >> Cell growth and death >> Apoptosis - multiple species
Cellular Processes >> Cell growth and death >> Necroptosis
Cellular Processes >> Cellular community - eukaryotes >> Focal adhesion
Organismal Systems >> Immune system >> NOD-like receptor signaling pathway
Human Diseases >> Cancer: overview >> Pathways in cancer
Human Diseases >> Cancer: specific types >> Small cell lung cancer
Environmental Information Processing >> Signal transduction >> NF-kappa B signaling pathway
Environmental Information Processing >> Signal transduction >> TNF signaling pathway
Genetic Information Processing >> Folding, sorting and degradation >> Ubiquitin mediated proteolysis
Reference Citation({{totalcount}})
Catalog: YP0681
Size
Price
Status
Qty.
200μL
$600.00
In stock
0
100μL
$340.00
In stock
0
50μL
$190.00
In stock
0
Add to cart
Collected
Collect
Recently Viewed Products
Clear allPRODUCTS
CUSTOMIZED
ABOUT US








Toggle night Mode
{{pinfoXq.title || ''}}
Catalog: {{pinfoXq.catalog || ''}}
Filter:
All
{{item.name}}
{{pinfo.title}}
-{{pinfo.catalog}}
Main Information
Target
{{pinfo.target}}
Reactivity
{{pinfo.react}}
Applications
{{pinfo.applicat}}
Conjugate/Modification
{{pinfo.coupling}}/{{pinfo.modific}}
MW (kDa)
{{pinfo.mwcalc}}
Host Species
{{pinfo.hostspec}}
Isotype
{{pinfo.isotype}}
Product {{index}}/{{pcount}}
Prev
Next
{{pvTitle}}
Scroll wheel zooms the picture
{{pvDescr}}