





Catalog: YN2471
Size
Price
Status
Qty.
200μL
$450.00
In stock
0
100μL
$280.00
In stock
0
40μL
$150.00
In stock
0
Add to cart


Collected
Collect
Main Information
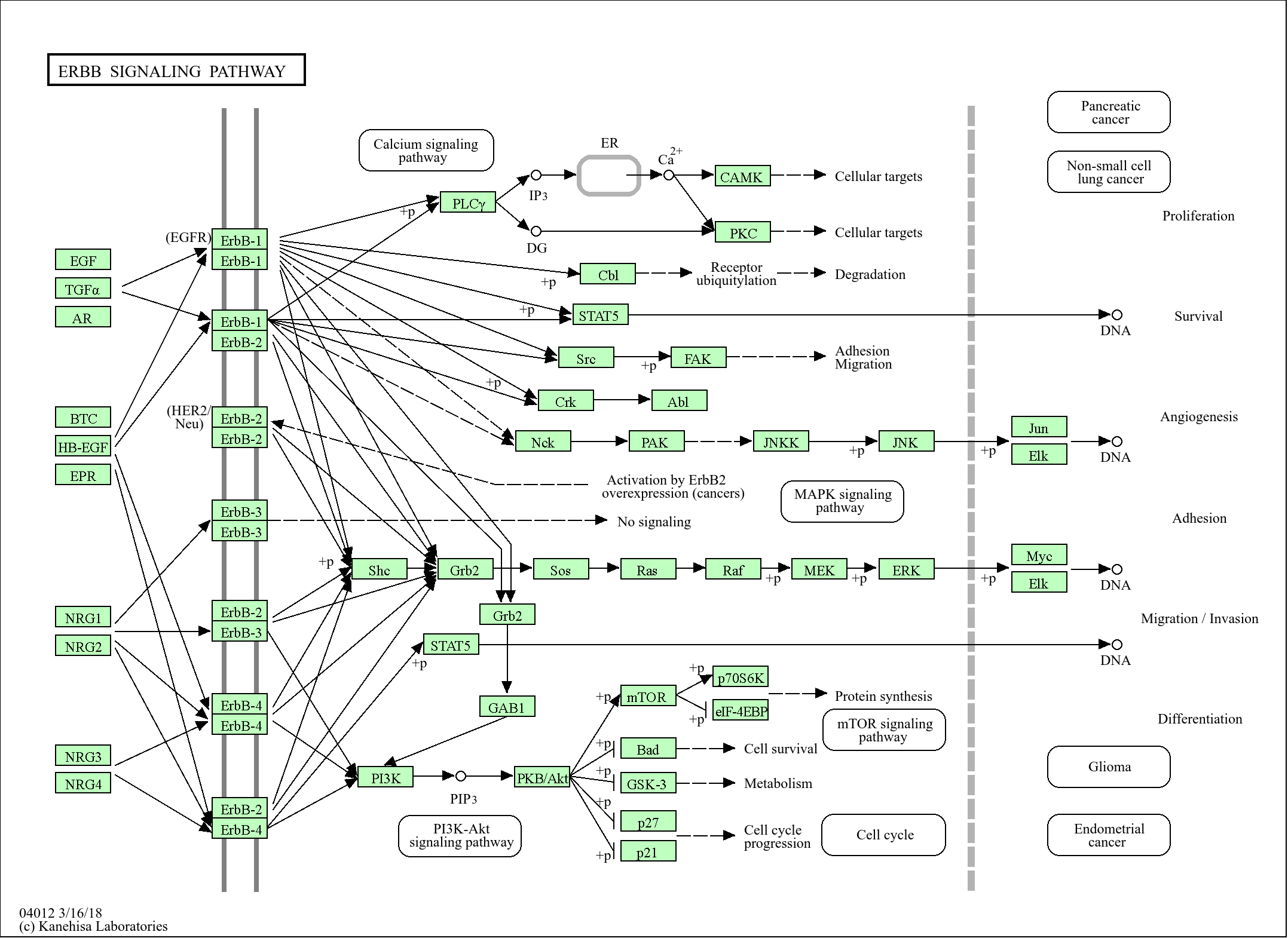
Target
CLN8
Host Species
Rabbit
Reactivity
Human, Rat, Mouse,
Applications
WB, ELISA
MW
31kD (Observed)
Conjugate/Modification
Unmodified
Detailed Information
Recommended Dilution Ratio
WB 1:500-2000; ELISA 1:5000-20000
Formulation
Liquid in PBS containing 50% glycerol,0.5% BSA and 0.02% sodium azide.
Specificity
CLN8 Polyclonal Antibody detects endogenous levels of protein.
Purification
The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen.
Storage
-15°C to -25°C/1 year(Do not lower than -25°C)
Concentration
1 mg/ml
MW(Observed)
31kD
Modification
Unmodified
Clonality
Polyclonal
Isotype
IgG
Related Products
Secondary Antibodies
Goat Anti Mouse IgG(H+L) (HRP)
RS0001
More→
Secondary Antibodies
Goat Anti Rabbit IgG(H+L) (HRP)
RS0002
More→
Primary Antibodies
β-actin (PTR2364) Mouse mAb
YM3028
More→
Primary Antibodies
GAPDH (PTR2304) Mouse mAb
YM3029
More→
Antigen&Target Information
Immunogen:
Synthesized peptide derived from human protein . at AA range: 231-280
show all
Specificity:
CLN8 Polyclonal Antibody detects endogenous levels of protein.
show all
Gene Name:
CLN8 C8orf61
show all
Protein Name:
Protein CLN8
show all
Background:
ceroid-lipofuscinosis, neuronal 8(CLN8) Homo sapiens This gene encodes a transmembrane protein belonging to a family of proteins containing TLC domains, which are postulated to function in lipid synthesis, transport, or sensing. The protein localizes to the endoplasmic reticulum (ER), and may recycle between the ER and ER-Golgi intermediate compartment. Mutations in this gene are associated with progressive epilepsy with mental retardation (EMPR), which is a subtype of neuronal ceroid lipofuscinoses (NCL). Patients with mutations in this gene have altered levels of sphingolipid and phospholipids in the brain. [provided by RefSeq, Jul 2008],
show all
Function:
Disease:Defects in CLN8 are the cause of neuronal ceroid lipofuscinosis 8 (CLN8) [MIM:600143]. Childhood-onset neuronal ceroid lipofuscinoses (NCL) are a group of autosomal recessive progressive encephalopathies characterized by the accumulation of autofluorescent material, mainly ATP synthase subunit C, in various tissues, notably in neurons. Based on clinical features, the country of origin of patients, and the molecular genetic background of the disorder, at least seven different forms are thought to exist. CLN8 is characterized by normal early development, onset of generalized seizures between 5 and 10 years, and subsequent progressive mental retardation.,Disease:Defects in CLN8 are the cause of progressive epilepsy with mental retardation (EPMR) [MIM:610003]; also called Northern epilepsy variant of neuronal ceroid lipofuscinosis 8. EPMR is a form of NCL so far described only in Finland. It has been considered as a distinct clinical and genetic entity among the NCL.,online information:Neural Ceroid Lipofuscinoses mutation db,PTM:Does not seem to be N-glycosylated.,similarity:Contains 1 TLC (TRAM/LAG1/CLN8) domain.,
show all
Cellular Localization:
Endoplasmic reticulum membrane ; Multi-pass membrane protein . Endoplasmic reticulum-Golgi intermediate compartment membrane ; Multi-pass membrane protein . Endoplasmic reticulum .
show all
Tissue Expression:
Placenta,Uterus,
show all
Reference Citation({{totalcount}})
Catalog: YN2471
Size
Price
Status
Qty.
200μL
$450.00
In stock
0
100μL
$280.00
In stock
0
40μL
$150.00
In stock
0
Add to cart
Collected
Collect
Recently Viewed Products
Clear allPRODUCTS
CUSTOMIZED
ABOUT US








Toggle night Mode
{{pinfoXq.title || ''}}
Catalog: {{pinfoXq.catalog || ''}}
Filter:
All
{{item.name}}
{{pinfo.title}}
-{{pinfo.catalog}}
Main Information
Target
{{pinfo.target}}
Reactivity
{{pinfo.react}}
Applications
{{pinfo.applicat}}
Conjugate/Modification
{{pinfo.coupling}}/{{pinfo.modific}}
MW (kDa)
{{pinfo.mwcalc}}
Host Species
{{pinfo.hostspec}}
Isotype
{{pinfo.isotype}}
Product {{index}}/{{pcount}}
Prev
Next
{{pvTitle}}
Scroll wheel zooms the picture
{{pvDescr}}