





Catalog: YN2009
Size
Price
Status
Qty.
200μL
$450.00
In stock
0
100μL
$280.00
In stock
0
40μL
$150.00
In stock
0
Add to cart


Collected
Collect
Main Information
Target
PRIO
Host Species
Rabbit
Reactivity
Human, Rat, Mouse
Applications
WB, ELISA
MW
27kD (Observed)
Conjugate/Modification
Unmodified
Detailed Information
Recommended Dilution Ratio
WB 1:500-2000; ELISA 1:5000-20000
Formulation
Liquid in PBS containing 50% glycerol, and 0.02% sodium azide.
Specificity
PRIO Polyclonal Antibody detects endogenous levels of protein.
Purification
The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen.
Storage
-15°C to -25°C/1 year(Do not lower than -25°C)
Concentration
1 mg/ml
MW(Observed)
27kD
Modification
Unmodified
Clonality
Polyclonal
Isotype
IgG
Related Products
Secondary Antibodies
Goat Anti Mouse IgG(H+L) (HRP)
RS0001
More→
Secondary Antibodies
Goat Anti Rabbit IgG(H+L) (HRP)
RS0002
More→
Primary Antibodies
β-actin (PTR2364) Mouse mAb
YM3028
More→
Primary Antibodies
GAPDH (PTR2304) Mouse mAb
YM3029
More→
Antigen&Target Information
Immunogen:
Synthesized peptide derived from part region of human protein
show all
Specificity:
PRIO Polyclonal Antibody detects endogenous levels of protein.
show all
Gene Name:
PRNP PRIP PRP
show all
Protein Name:
Major prion protein (PrP) (ASCR) (PrP27-30) (PrP33-35C) (CD antigen CD230)
show all
Background:
The protein encoded by this gene is a membrane glycosylphosphatidylinositol-anchored glycoprotein that tends to aggregate into rod-like structures. The encoded protein contains a highly unstable region of five tandem octapeptide repeats. This gene is found on chromosome 20, approximately 20 kbp upstream of a gene which encodes a biochemically and structurally similar protein to the one encoded by this gene. Mutations in the repeat region as well as elsewhere in this gene have been associated with Creutzfeldt-Jakob disease, fatal familial insomnia, Gerstmann-Straussler disease, Huntington disease-like 1, and kuru. An overlapping open reading frame has been found for this gene that encodes a smaller, structurally unrelated protein, AltPrp. Alternative splicing results in multiple transcript variants. [provided by RefSeq, Nov 2014],
show all
Function:
Disease:Defects in PRNP are the cause of Creutzfeldt-Jakob disease (CJD) [MIM:123400]. CJD occurs primarily as a sporadic disorder (1 per million), while 10-15% are familial. Accidental transmission of CJD to humans appears to be iatrogenic (contaminated human growth hormone (HGH), corneal transplantation, electroencephalographic electrode implantation, etc.). Epidemiologic studies have failed to implicate the ingestion of infected annimal meat in the pathogenesis of CJD in human. The triad of microscopic features that characterize the prion diseases consists of (1) spongiform degeneration of neurons, (2) severe astrocytic gliosis that often appears to be out of proportion to the degree of nerve cell loss, and (3) amyloid plaque formation. CJD is characterized by progressive dementia and myoclonic seizures, affecting adults in mid-life. Some patients present sleep disorders, abnormalities of high cortical function, cerebellar and corticospinal disturbances. The disease ends in death after a 3-12 months illness.,Disease:Defects in PRNP are the cause of fatal familial insomnia (FFI) [MIM:600072]. FFI is an autosomal dominant disorder and is characterized by neuronal degeneration limited to selected thalamic nuclei and progressive insomnia.,Disease:Defects in PRNP are the cause of Gerstmann-Straussler disease (GSD) [MIM:137440]. GSD is a heterogeneous disorder and was defined as a spinocerebellar ataxia with dementia and plaquelike deposits. GSD incidence is less than 2 per 100 million live births.,Disease:Defects in PRNP are the cause of Huntington disease-like 1 (HDL1) [MIM:603218]. HDL1 is an autosomal dominant, early onset neurodegenerative disorder with prominent psychiatric features.,Disease:Defects in PRNP are the cause of kuru [MIM:245300]. Kuru is transmitted during ritualistic cannibalism, among natives of the New Guinea highlands. Patients exhibit various movement disorders like cerebellar abnormalities, rigidity of the limbs, and clonus. Emotional lability is present, and dementia is conspicuously absent. Death usually occurs from 3 to 12 month after onset.,Disease:Defects in PRNP are the cause of prion disease with protracted course [MIM:606688]; an autosomal dominant presenile dementia with a rapidly progressive and protracted clinical course. The dementia was characterized clinically by frontotemporal features, including early personality changes. Some patients had memory loss, several showed aggressiveness, hyperorality and verbal stereotypy, others had parkinsonian symptoms.,Disease:PrP is found in high quantity in the brain of humans and animals infected with neurodegenerative diseases known as transmissible spongiform encephalopathies or prion diseases, like: Creutzfeldt-Jakob disease (CJD), fatal familial insomnia (FFI), Gerstmann-Straussler disease (GSD), Huntington disease-like 1 (HDL1) and kuru in humans; scrapie in sheep and goat; bovine spongiform encephalopathy (BSE) in cattle; transmissible mink encephalopathy (TME); chronic wasting disease (CWD) of mule deer and elk; feline spongiform encephalopathy (FSE) in cats and exotic ungulate encephalopathy (EUE) in nyala and greater kudu. The prion diseases illustrate three manifestations of CNS degeneration: (1) infectious (2) sporadic and (3) dominantly inherited forms. TME, CWD, BSE, FSE, EUE are all thought to occur after consumption of prion-infected foodstuffs.,Function:The physiological function of PrP is not known.,online information:PRNP entry,polymorphism:The five tandem octapeptide repeats region is highly unstable. Insertions or deletions of octapeptide repeat units are associated to prion disease.,PTM:The glycosylation pattern (the amount of mono-, di- and non-glycosylated forms or glycoforms) seems to differ in normal and CJD prion.,similarity:Belongs to the prion family.,subunit:PrP has a tendency to aggregate yielding polymers called "rods". Interacts with GRB2, PRNPIP and SYN1.,
show all
Cellular Localization:
Cell membrane; Lipid-anchor, GPI-anchor . Golgi apparatus . Targeted to lipid rafts via association with the heparan sulfate chains of GPC1. Colocates, in the presence of Cu(2+), to vesicles in para- and perinuclear regions, where both proteins undergo internalization. Heparin displaces PRNP from lipid rafts and promotes endocytosis. .
show all
Tissue Expression:
Blood,Brain,Ovary,Prostate,
show all
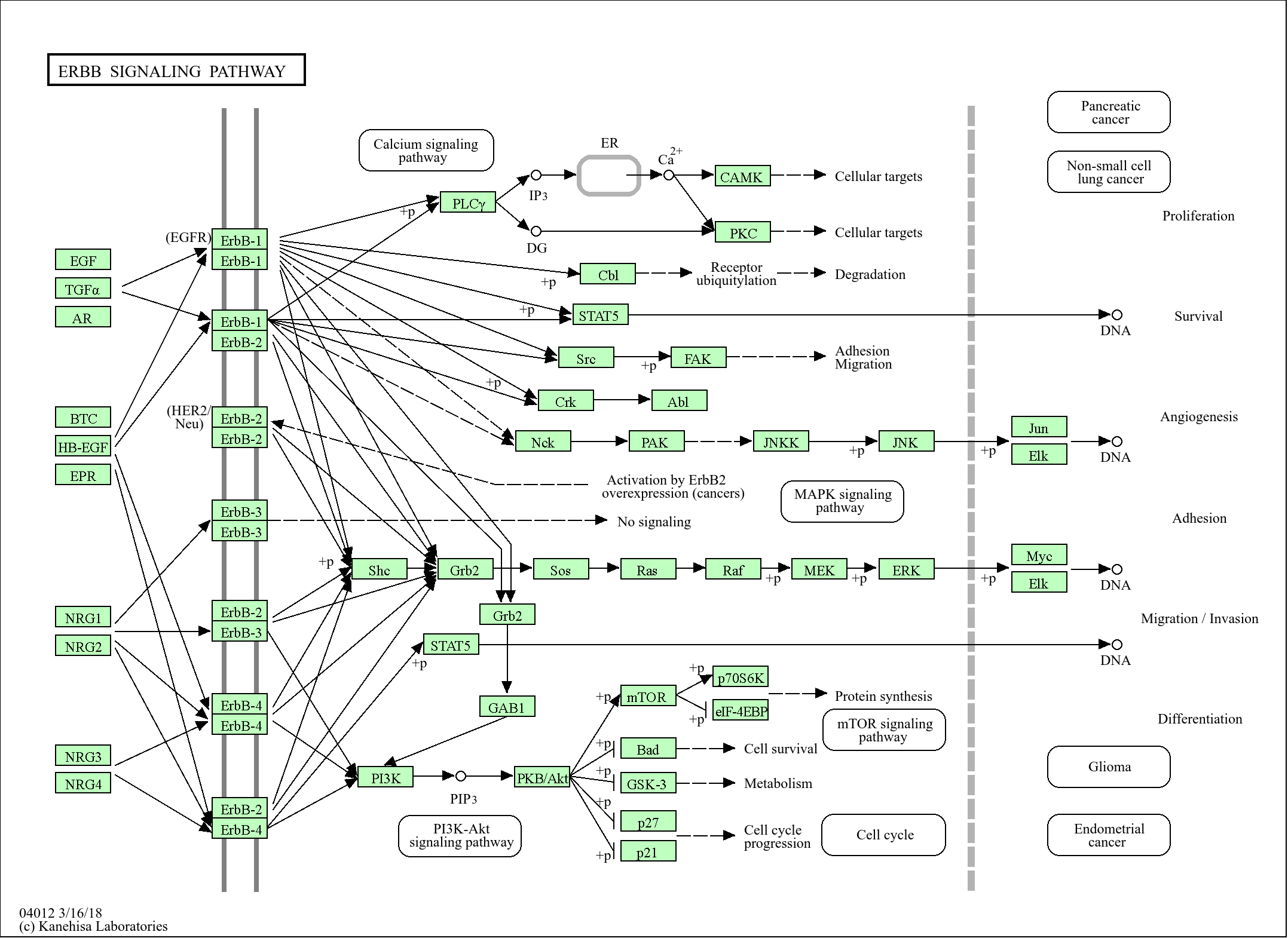
Research Areas:
>>Ferroptosis ;
>>Prion disease ;
>>Pathways of neurodegeneration - multiple diseases
>>Prion disease ;
>>Pathways of neurodegeneration - multiple diseases
show all
Signaling Pathway
Reference Citation({{totalcount}})
Catalog: YN2009
Size
Price
Status
Qty.
200μL
$450.00
In stock
0
100μL
$280.00
In stock
0
40μL
$150.00
In stock
0
Add to cart
Collected
Collect
Recently Viewed Products
Clear allPRODUCTS
CUSTOMIZED
ABOUT US








Toggle night Mode
{{pinfoXq.title || ''}}
Catalog: {{pinfoXq.catalog || ''}}
Filter:
All
{{item.name}}
{{pinfo.title}}
-{{pinfo.catalog}}
Main Information
Target
{{pinfo.target}}
Reactivity
{{pinfo.react}}
Applications
{{pinfo.applicat}}
Conjugate/Modification
{{pinfo.coupling}}/{{pinfo.modific}}
MW (kDa)
{{pinfo.mwcalc}}
Host Species
{{pinfo.hostspec}}
Isotype
{{pinfo.isotype}}
Product {{index}}/{{pcount}}
Prev
Next
{{pvTitle}}
Scroll wheel zooms the picture
{{pvDescr}}