



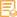


Catalog: YP1488
Size
Price
Status
Qty.
200μL
$600.00
In stock
0
100μL
$340.00
In stock
0
50μL
$190.00
In stock
0
Add to cart


Collected
Collect
Main Information
Target
Shc Phospho Ser239/240
Host Species
Rabbit
Reactivity
Human, Mouse, Rat
Applications
WB, ELISA, IHC
MW
66kD(p66),52kD(p52),46kD(p46)kD (Observed)
Conjugate/Modification
phosphate
Detailed Information
Recommended Dilution Ratio
WB 1:500-2000; IHC 1:50-300; ELISA 1:2000-20000
Formulation
Liquid in PBS containing 50% glycerol, 0.5% BSA and 0.02% sodium azide.
Specificity
This antibody detects endogenous levels of Shc only when phosphorylated at Ser239 or ser240,and dually phosphorylated at two sites.
Purification
The antibody was affinity-purified from rabbit serum by affinity-chromatography using specific immunogen.
Storage
-15°C to -25°C/1 year(Do not lower than -25°C)
Concentration
1 mg/ml
MW(Observed)
66kD(p66),52kD(p52),46kD(p46)kD
Modification
phosphate
Clonality
Polyclonal
Isotype
IgG
Related Products
Primary Antibodies

Shc Rabbit pAb
YT4287
More→
Primary Antibodies
Shc (Phospho Ser36) Rabbit pAb
YP1075
More→
Primary Antibodies
Shc (Phospho Tyr427) Rabbit pAb
YP0579
More→
Primary Antibodies
Shc (Phospho Tyr349) Rabbit pAb
YP0578
More→
ELISA Kits
Total Shc (Ab-349) Cell-Based Colorimetric ELISA Kit
KA4279C
More→
ELISA Kits
Shc (Phospho Tyr427) Cell-Based Colorimetric ELISA Kit
KA1179C
More→
ELISA Kits
Shc (Phospho Tyr349) Cell-Based Colorimetric ELISA Kit
KA1178C
More→
Antigen&Target Information
Immunogen:
Synthesized phosho peptide around human Shc (Ser239 and 240)
show all
Specificity:
This antibody detects endogenous levels of Shc only when phosphorylated at Ser239 or ser240,and dually phosphorylated at two sites.
show all
Gene Name:
SHC1 SHC SHCA
show all
Protein Name:
Shc (Ser239/240)
show all
Other Name:
SHC-transforming protein 1 ;
SHC-transforming protein 3 ;
SHC-transforming protein A ;
Src homology 2 domain-containing-transforming protein C1 ;
SH2 domain protein C1 ;
SHC-transforming protein 3 ;
SHC-transforming protein A ;
Src homology 2 domain-containing-transforming protein C1 ;
SH2 domain protein C1 ;
show all
Database Link:
Background:
This gene encodes three main isoforms that differ in activities and subcellular location. While all three are adapter proteins in signal transduction pathways, the longest (p66Shc) may be involved in regulating life span and the effects of reactive oxygen species. The other two isoforms, p52Shc and p46Shc, link activated receptor tyrosine kinases to the Ras pathway by recruitment of the GRB2/SOS complex. p66Shc is not involved in Ras activation. Unlike the other two isoforms, p46Shc is targeted to the mitochondrial matrix. Several transcript variants encoding different isoforms have been found for this gene. [provided by RefSeq, Feb 2011],
show all
Function:
Domain:In response to a variety of growth factors, isoform p46Shc and isoform p52Shc bind to phosphorylated Trk receptors through their phosphotyrosine binding (PID) and/or SH2 domains. The PID and SH2 domains bind to specific phosphorylated tyrosine residues in the Asn-Pro-Xaa-Tyr(P) motif of the Trk receptors. Isoform p46Shc and isoform p52Shc are in turn phosphorylated on three tyrosine residues within the extended proline-rich domain. These phosphotyrosines act as docking site for GRB2 and thereby are involved in Ras activation.,Function:Signaling adapter that couples activated growth factor receptors to signaling pathway. Isoform p46Shc and isoform p52Shc, once phosphorylated, couple activated receptor tyrosine kinases to Ras via the recruitment of the GRB2/SOS complex and are implicated in the cytoplasmic propagation of mitogenic signals. Isoform p46Shc and isoform p52Shc may thus function as initiators of the Ras signaling cascade in various non-neuronal systems. Isoform p66Shc does not mediate Ras activation, but is involved in signal transduction pathways that regulate the cellular response to oxidative stress and life span. Isoform p66Shc acts as a downstream target of the tumor suppressor p53 and is indispensable for the ability of stress-activated p53 to induce elevation of intracellular oxidants, cytochrome c release and apoptosis. The expression of isoform p66Shc has been correlated with life span.,PTM:Phosphorylated by activated epidermal growth factor receptor. Isoform p46Shc and isoform p52Shc are phosphorylated on tyrosine residues of the Pro-rich domain. Isoform p66Shc is phosphorylated on Ser-36 upon treatment with insulin, hydrogen peroxide or irradiation with ultraviolet light.,similarity:Contains 1 PID domain.,similarity:Contains 1 SH2 domain.,subcellular location:Localized to the mitochondria matrix. Targeting of isoform p46Shc to mitochondria is mediated by its first 32 amino acids, which behave as a bona fide mitochondrial targeting sequence. Isoform p52Shc and isoform p66Shc, that contain the same sequence but more internally located, display a different subcellular localization.,subunit:Interacts with the Trk receptors in a phosphotyrosine-dependent manner. Interacts with the NPXY motif of tyrosine-phosphorylated IGF1R and INSR in vitro via the PID domain. Once activated, binds to GRB2. Interacts with tyrosine-phosphorylated CD3T. Interacts with the N-terminal region of APS. Interacts with phosphorylated LRP1 and IRS4. Interacts with INPP5D/SHIP1 and INPPL1/SHIP2.,tissue specificity:Widely expressed. Expressed in neural stem cells but absent in mature neurons.,
show all
Cellular Localization:
Cytoplasm.; [Isoform p46Shc]: Mitochondrion matrix . Localized to the mitochondria matrix. Targeting of isoform p46Shc to mitochondria is mediated by its first 32 amino acids, which behave as a bona fide mitochondrial targeting sequence. Isoform p52Shc and isoform p66Shc, that contain the same sequence but more internally located, display a different subcellular localization.; [Isoform p66Shc]: Mitochondrion . In case of oxidative conditions, phosphorylation at 'Ser-36' of isoform p66Shc, leads to mitochondrial accumulation. .
show all
Tissue Expression:
Research Areas:
>>EGFR tyrosine kinase inhibitor resistance ;
>>Endocrine resistance ;
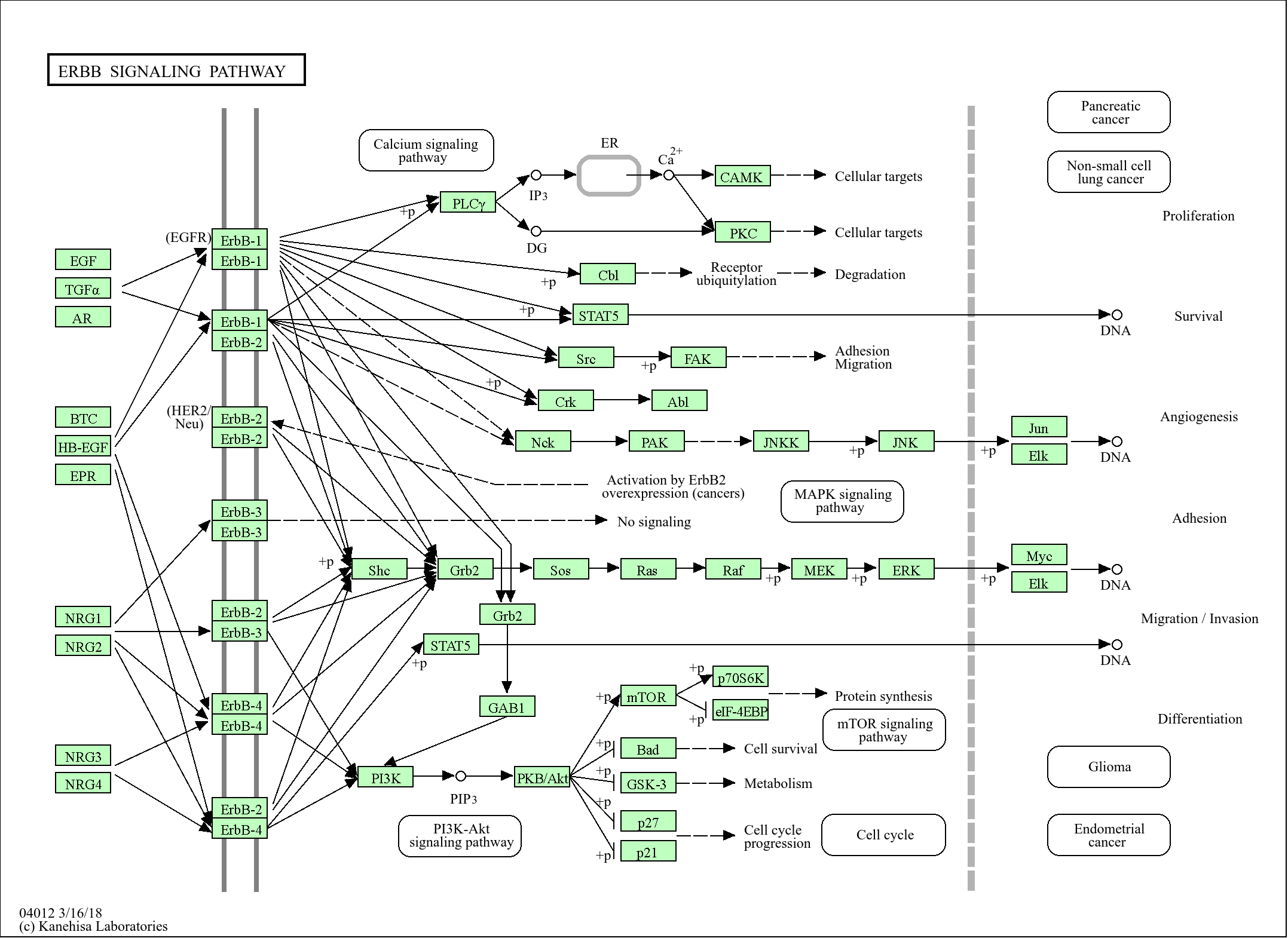
>>ErbB signaling pathway ;
>>Ras signaling pathway ;
>>Chemokine signaling pathway ;
>>Phospholipase D signaling pathway ;
>>Focal adhesion ;
>>Natural killer cell mediated cytotoxicity ;
>>Neurotrophin signaling pathway ;
>>Insulin signaling pathway ;
>>Estrogen signaling pathway ;
>>Prolactin signaling pathway ;
>>Relaxin signaling pathway ;
>>Growth hormone synthesis, secretion and action ;
>>Alcoholism ;
>>Bacterial invasion of epithelial cells ;
>>MicroRNAs in cancer ;
>>Glioma ;
>>Chronic myeloid leukemia ;
>>Breast cancer ;
>>Hepatocellular carcinoma ;
>>Gastric cancer
>>Endocrine resistance ;
>>ErbB signaling pathway ;
>>Ras signaling pathway ;
>>Chemokine signaling pathway ;
>>Phospholipase D signaling pathway ;
>>Focal adhesion ;
>>Natural killer cell mediated cytotoxicity ;
>>Neurotrophin signaling pathway ;
>>Insulin signaling pathway ;
>>Estrogen signaling pathway ;
>>Prolactin signaling pathway ;
>>Relaxin signaling pathway ;
>>Growth hormone synthesis, secretion and action ;
>>Alcoholism ;
>>Bacterial invasion of epithelial cells ;
>>MicroRNAs in cancer ;
>>Glioma ;
>>Chronic myeloid leukemia ;
>>Breast cancer ;
>>Hepatocellular carcinoma ;
>>Gastric cancer
show all
Signaling Pathway
Cellular Processes >> Cellular community - eukaryotes >> Focal adhesion
Organismal Systems >> Immune system >> Natural killer cell mediated cytotoxicity
Organismal Systems >> Immune system >> Chemokine signaling pathway
Organismal Systems >> Endocrine system >> Insulin signaling pathway
Organismal Systems >> Endocrine system >> Estrogen signaling pathway
Organismal Systems >> Endocrine system >> Prolactin signaling pathway
Organismal Systems >> Endocrine system >> Relaxin signaling pathway
Organismal Systems >> Endocrine system >> Growth hormone synthesis, secretion and action
Organismal Systems >> Nervous system >> Neurotrophin signaling pathway
Human Diseases >> Cancer: overview >> MicroRNAs in cancer
Human Diseases >> Cancer: specific types >> Hepatocellular carcinoma
Human Diseases >> Cancer: specific types >> Gastric cancer
Human Diseases >> Cancer: specific types >> Glioma
Human Diseases >> Cancer: specific types >> Chronic myeloid leukemia
Human Diseases >> Cancer: specific types >> Breast cancer
Environmental Information Processing >> Signal transduction >> ErbB signaling pathway
Environmental Information Processing >> Signal transduction >> Ras signaling pathway
Environmental Information Processing >> Signal transduction >> Phospholipase D signaling pathway
Reference Citation({{totalcount}})
Catalog: YP1488
Size
Price
Status
Qty.
200μL
$600.00
In stock
0
100μL
$340.00
In stock
0
50μL
$190.00
In stock
0
Add to cart
Collected
Collect
Recently Viewed Products
Clear allPRODUCTS
CUSTOMIZED
ABOUT US








Toggle night Mode
{{pinfoXq.title || ''}}
Catalog: {{pinfoXq.catalog || ''}}
Filter:
All
{{item.name}}
{{pinfo.title}}
-{{pinfo.catalog}}
Main Information
Target
{{pinfo.target}}
Reactivity
{{pinfo.react}}
Applications
{{pinfo.applicat}}
Conjugate/Modification
{{pinfo.coupling}}/{{pinfo.modific}}
MW (kDa)
{{pinfo.mwcalc}}
Host Species
{{pinfo.hostspec}}
Isotype
{{pinfo.isotype}}
Product {{index}}/{{pcount}}
Prev
Next
{{pvTitle}}
Scroll wheel zooms the picture
{{pvDescr}}