





Catalog: YT4915
Size
Price
Status
Qty.
200μL
$450.00
In stock
0
100μL
$280.00
In stock
0
40μL
$150.00
In stock
0
Add to cart


Collected
Collect
Main Information
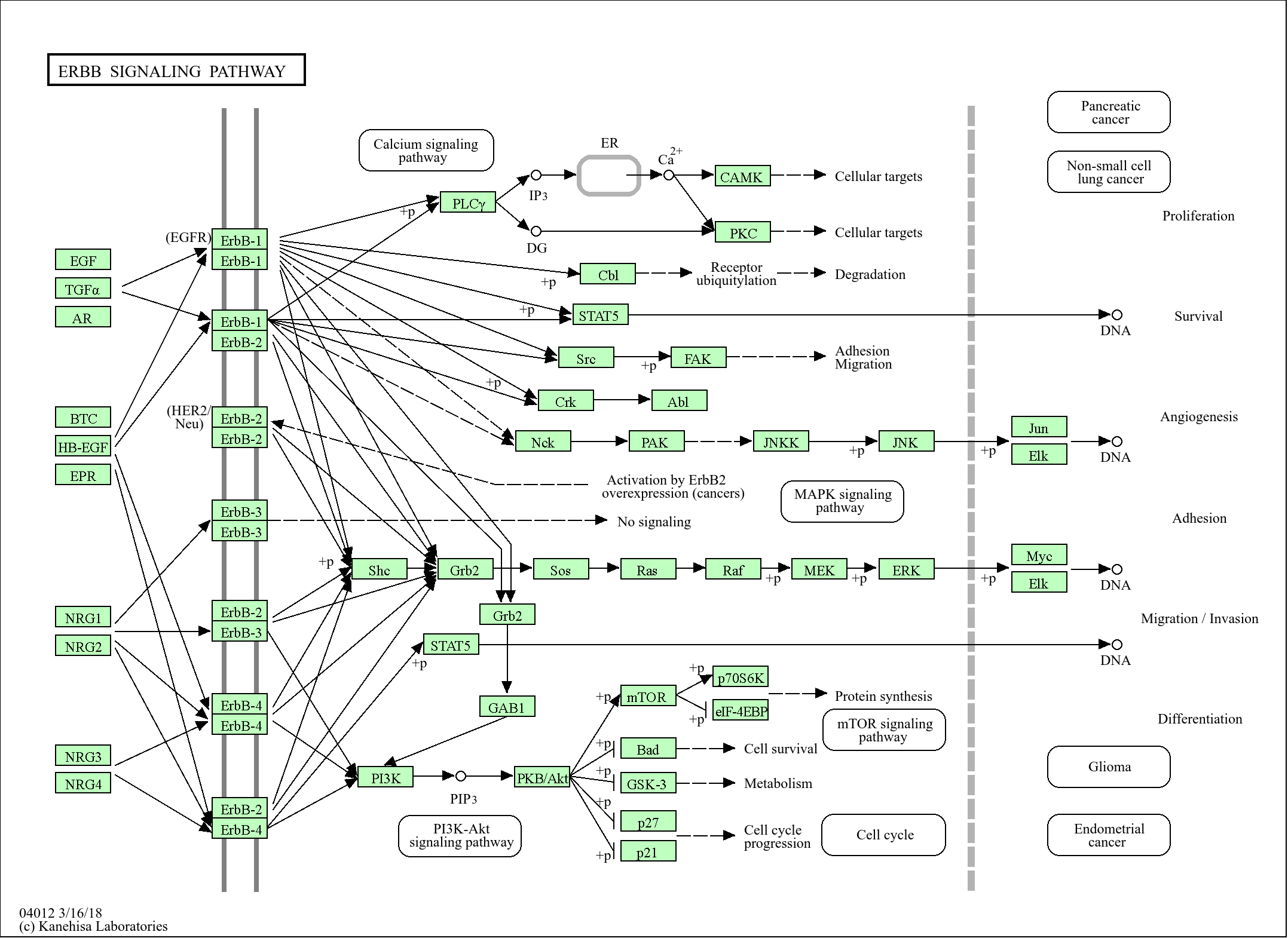
Target
XPG
Host Species
Rabbit
Reactivity
Human, Mouse
Applications
WB, IHC, IF, ELISA
MW
130kD (Observed)
Conjugate/Modification
Unmodified
Detailed Information
Recommended Dilution Ratio
WB 1:500-1:2000; IHC 1:100-1:300; IF 1:200-1:1000; ELISA 1:5000; Not yet tested in other applications.
Formulation
Liquid in PBS containing 50% glycerol, 0.5% BSA and 0.02% sodium azide.
Specificity
XPG Polyclonal Antibody detects endogenous levels of XPG protein.
Purification
The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen.
Storage
-15°C to -25°C/1 year(Do not lower than -25°C)
Concentration
1 mg/ml
MW(Observed)
130kD
Modification
Unmodified
Clonality
Polyclonal
Isotype
IgG
Related Products
Primary Antibodies

XPG Rabbit pAb
YT4915
More→
ELISA Kits

Total XPG Cell-Based Colorimetric ELISA Kit
KA3579C
More→
Secondary Antibodies
Goat Anti Mouse IgG(H+L) (HRP)
RS0001
More→
Secondary Antibodies
Goat Anti Rabbit IgG(H+L) (HRP)
RS0002
More→
Primary Antibodies
β-actin (PTR2364) Mouse mAb
YM3028
More→
Primary Antibodies
GAPDH (PTR2304) Mouse mAb
YM3029
More→
Antigen&Target Information
Immunogen:
The antiserum was produced against synthesized peptide derived from human ERCC5. AA range:131-180
show all
Specificity:
XPG Polyclonal Antibody detects endogenous levels of XPG protein.
show all
Gene Name:
ERCC5
show all
Protein Name:
DNA repair protein complementing XP-G cells
show all
Other Name:
ERCC5 ;
ERCM2 ;
XPG ;
XPGC ;
DNA repair protein complementing XP-G cells ;
DNA excision repair protein ERCC-5 ;
Xeroderma pigmentosum group G-complementing protein
ERCM2 ;
XPG ;
XPGC ;
DNA repair protein complementing XP-G cells ;
DNA excision repair protein ERCC-5 ;
Xeroderma pigmentosum group G-complementing protein
show all
Background:
This gene encodes a single-strand specific DNA endonuclease that makes the 3' incision in DNA excision repair following UV-induced damage. The protein may also function in other cellular processes, including RNA polymerase II transcription, and transcription-coupled DNA repair. Mutations in this gene cause xeroderma pigmentosum complementation group G (XP-G), which is also referred to as xeroderma pigmentosum VII (XP7), a skin disorder characterized by hypersensitivity to UV light and increased susceptibility for skin cancer development following UV exposure. Some patients also develop Cockayne syndrome, which is characterized by severe growth defects, mental retardation, and cachexia. Read-through transcription exists between this gene and the neighboring upstream BIVM (basic, immunoglobulin-like variable motif containing) gene. [provided by RefSeq, Feb 2011],
show all
Function:
cofactor:Binds 2 magnesium ions per subunit. They probably participate in the reaction catalyzed by the enzyme. May bind an additional third magnesium ion after substrate binding.,Disease:Defects in ERCC5 are the cause of xeroderma pigmentosum complementation group G (XP-G) [MIM:278780]; also known as xeroderma pigmentosum VII (XP7). Xeroderma pigmentosum is an autosomal recessive pigmentary skin disorder characterized by solar hypersensitivity of the skin, high predisposition for developing cancers on areas exposed to sunlight and, in some cases, neurological abnormalities. Some XP-G patients present features of Cockayne syndrome, including dwarfism, sensorineural deafness, microcephaly, mental retardation, pigmentary retinopathy, ataxia, decreased nerve conduction velocities.,Function:Single-stranded structure-specific DNA endonuclease involved in DNA excision repair. Makes the 3'incision in DNA nucleotide excision repair (NER). Acts as a cofactor for a DNA glycosylase that removes oxidized pyrimidines from DNA. May also be involved in transcription-coupled repair of this kind of damage, in transcription by RNA polymerase II, and perhaps in other processes too.,similarity:Belongs to the XPG/RAD2 endonuclease family. XPG subfamily.,subunit:Interacts with PCNA.,
show all
Cellular Localization:
Nucleus . Chromosome . Colocalizes with RAD51 to nuclear foci in S phase (PubMed:26833090). Localizes to DNA double-strand breaks (DBS) during replication stress (PubMed:26833090). Colocalizes with BRCA2 to nuclear foci following DNA replication stress (PubMed:26833090). .
show all
Tissue Expression:
Bone marrow,Epithelium,Eye,
show all
Research Areas:
>>Nucleotide excision repair
show all
Reference Citation({{totalcount}})
Catalog: YT4915
Size
Price
Status
Qty.
200μL
$450.00
In stock
0
100μL
$280.00
In stock
0
40μL
$150.00
In stock
0
Add to cart
Collected
Collect
Recently Viewed Products
Clear allPRODUCTS
CUSTOMIZED
ABOUT US








Toggle night Mode
{{pinfoXq.title || ''}}
Catalog: {{pinfoXq.catalog || ''}}
Filter:
All
{{item.name}}
{{pinfo.title}}
-{{pinfo.catalog}}
Main Information
Target
{{pinfo.target}}
Reactivity
{{pinfo.react}}
Applications
{{pinfo.applicat}}
Conjugate/Modification
{{pinfo.coupling}}/{{pinfo.modific}}
MW (kDa)
{{pinfo.mwcalc}}
Host Species
{{pinfo.hostspec}}
Isotype
{{pinfo.isotype}}
Product {{index}}/{{pcount}}
Prev
Next
{{pvTitle}}
Scroll wheel zooms the picture
{{pvDescr}}